
�������: 1-14 ���鵽��ԭ�ӷ�������ѧ ��̬����ؼ�¼14�� . ��ѯʱ��(0.07 ��)
��ѧ���״η���ԭ�Ӻ���̬���ڷ����ͽṹ
ԭ�Ӻ� ��̬ �����ͽṹ
<
2023/12/12
�й���ѧԺ���������о���������Ա�������߽����״�ͨ��ʵ��֤ʵ��ԭ�Ӻ���̬�д��ڷ����ͽṹ�����о���2023��11��21�շ����ڹ�������ѧ�ڿ����������ۿ챨���ϡ�
���ö�ο���̬����ã�MRCI+Q������,�����ࣨSbS��������͵�3��?-S��⼫�����е���̬�Լ���������-������ЧӦ��������õĦ�̬�����˼��㡣�õ�27��?-S����̬��������͵�12����̬�ĵ��ӽṹ�����׳��������ܼ�����Ϣ��Sbԭ�Ӻ�Sԭ���ܼ��ļ���ֵ��ʵ��ֵ����ܺá�������������-������ЧӦ�Թ��׳��������ܼ���Ӱ�������ϲ�����������X��3/2����X��1/2��,2��1/2����X��...
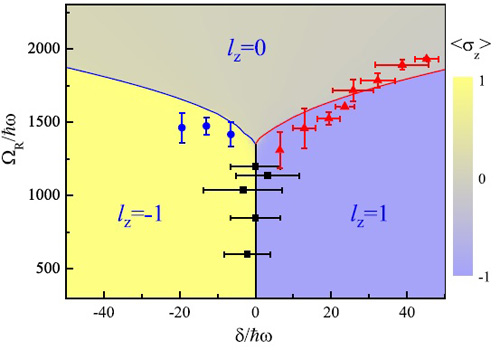
�й���ѧԺ�人��������ѧ�о����ȴ�ʵ���ϻ������-����Ƕ�����ϳ���ԭ����ϵ����̬��ͼ��ͼ��
�й���ѧԺ�人��������ѧ�о��� ����-��� �Ƕ��� ��� ����ԭ����ϵ ��̬��ͼ
<
2019/3/25
���գ��й���ѧԺ�人��������ѧ�о����о�Ա�������Ŷ�ʵ���˳���ԭ�ӵ�����-����Ƕ������ЧӦ���״δ�ʵ���ϻ���˸������ϵ����̬��ͼ���о��ɹ��������������������ۿ챨����(Phys. Rev. Lett. 122, 110402 (2019))���ù����Ǻ��ൺ��ѧ��������˹��ѧ�Լ��Ĵ�����˹�����Ƽ���ѧ�Ŀ�����Աһ�������ɵġ�
������ѧ������ѧ�μ������µ���� ��ԭ����̬��
����˫����������ϵ��ģ�Ͳ������ȫ��������ķ������о���AMF3(A=K, Rb; M=Zn, Cd, Ca):Ni2+��K2ZnF4:Ni2+��ϵ��Ni2+����̬���Ѻ;���ṹ�� ͨ��ģ����͵���˳�Ź���(EPR)�ף������ʾ���о���������Ϸ��ӵ��ܼ���ϸ�ṹ�;���ṹ����ʱ������F-����ϵ�����������ϻ�����Ӱ�첻�ɺ��ԣ�ͬʱ������EPR������нǡ���ѡ�����ϡ�ƽ�������Լ�ƫ������ı仯...
��̬����ԭ��(Z=2-54)������������ֵ��
���忼����H+2��̬���ƽ�������������H+2=1/2(1+S)1/2��H(z,ra)+��H(z,rb))�����ܼ���Ӱ�죬�õ��˻��ڴ˲������IJ���-�±���Ĭ�����µ�H+2�������ߵĽ�������ʽ�����ɴ˼���������.������֣���������H+2����̬����z��ȡ��1.23��ʱ�����ܼ���ƽ��λ�ö���ʵ��ֵ�����.�����ǽϸߵ����ܼ�ʱ���������еĪ�z��ȡ��1.13��������������ʱ�����ܼ���ʵ�����ܼ�...
SiH, SiD,SiT������̬(X2��)�Ľṹ��������ܺ���
SiH,SiD,SiT��̬ ���ӽṹ�����ܺ��� Murrell-Sorbie����
<
2009/10/29
����Ⱥ�ۼ�ԭ�ӷ��ӷ�Ӧ����ѧ���й�ԭ�����Ƶ���SiH(SiD,SiT)������̬�ĵ���̬�ͺ�������⼫�ޣ�����������ѧ��ͷ�㷨,Ӧ�ö�����̬�����QCISD/6-311g(df,2pd)������SiH,SiD,SiT����̬ƽ��ṹ��г��Ƶ�ʽ������Ż����㣮��ʹ�ø÷����ͻ����SiH(SiD,SiT)���ӵ���̬�����˵�����ɨ����㣬�����淽���������Murrel-Sorbie���ܺ������õ��˸�̬������...
Ӧ��MP2��CIS�����ֱ��Ż���IrR(CO)(PH3)2(mnt) [mnt��maleonitriledithiolate; R��H (1), CH3 (2), Br (3)]ϵ����������̬�ͼ���̬���νṹ. ʹ��TD-DFT�������������������պͷ������. ����������: �����1��3��430, 435��439 nm������������վ�ΪILCT/LLCT/MLCT���ԾǨ����, ���ǵ������...
����˫���Ӽ���K2��1��g��λ̬�����÷���ӫ����������о���1��g-3��g�����ײת�ƽ��档�ڴ�Kʵ���У����¿�����553��603 K֮�䣬Kԭ���ܶ��ɹ�ѧ���շ������õ���̽��1��g-11��+u��ֱ��ʱ��ֱ�ӫ��Ĺ�ǿ������һ����ָ��˥�����ߣ��ɴ˵õ�1��+g̬����Ч��������Ч�����ĵ�����K�ܶȳ����Թ�ϵ����ֱ�ߵ�б�ʵõ�1��g̬��������Ϊ(2.5��0.3)��10-14 cm2���ӽؾ�õ�����...
��ַ��ڼ�������������̬��ԭ�������е�Ӧ��
<
2007/12/12
ժҪ: ����̬��ԭ�ӵIJ������������Ĵ�ͳ���㷽������ǡ���������ۣ��������̵���ǡ����ʮ�ַ��ѣ�������������ݱ����ƽ��ɽ���ʽҲ�Ƚϸ��ӡ����ñ�ַ����Խ���ʽ����ʽ�������ۣ����������бȽϷ��㣬�������Ҳ��ʵ�����������
HF������̬(X1��+)�ķ��ӽṹ�����ܺ���
HF���� ��̬ ���ӽṹ ���ܺ���
<
2012/11/12
����Gaussian�����е����������巽��CCSD(T)��QCISD(T),�ֱ������6-311 + + G**��cc-pvdz���,�Ż�������HF������̬��ƽ��ṹ�������.���ñ�Murrell��Sorbie����,���з�������С���˷����,�õ���HF�������ܺ����Ľ�������ʽ,����һ���������HF���ӵ��������Լ����׳���.��������ʵ�����ݷdz��Ǻ�.
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...